|

I. Cerebral
protection and resuscitation
 Cerebral
protection. Cerebral protection is the
preemptive use of therapeutic interventions to
improve neurologic outcome in patients who will be
at risk for cerebral ischemia. The primary objective
is prevention of the deleterious effects of
ischemia.
Cerebral
protection. Cerebral protection is the
preemptive use of therapeutic interventions to
improve neurologic outcome in patients who will be
at risk for cerebral ischemia. The primary objective
is prevention of the deleterious effects of
ischemia.
Resuscitation.
Resuscitation refers to therapeutic interventions
initiated after an ischemic event. The goal is
treatment of ischemia and attenuation of neuronal
injury.
II. The ischemic
brain
 Cerebral ischemia. Cerebral ischemia is defined as
perfusion insufficient to provide the supply of
oxygen and nutrients needed for maintenance of
neuronal metabolic integrity (40% to 45% of total
cerebral metabolic rate for oxygen consumption
[CMRo2]) and function (55% to 60% of CMRo2). It is
assumed that a hierarchy of ischemic damage exists
in which neuronal function is abolished before
cellular integrity is lost.
Cerebral ischemia. Cerebral ischemia is defined as
perfusion insufficient to provide the supply of
oxygen and nutrients needed for maintenance of
neuronal metabolic integrity (40% to 45% of total
cerebral metabolic rate for oxygen consumption
[CMRo2]) and function (55% to 60% of CMRo2). It is
assumed that a hierarchy of ischemic damage exists
in which neuronal function is abolished before
cellular integrity is lost.
Cellular integrity. The brain utilizes glucose as
its primary substrate for energy production. In the nonfasting state, glucose is metabolized via
oxidative phosphorylation to adenosine triphosphate
(ATP), which is needed for cellular activities such
as homeostasis, protein synthesis, removal of carbon
dioxide (CO2), mitochondrial activity, and
maintenance of ionic gradients and cell membrane
stability.
Neuronal function. Normal neuronal functional
activity consists of the generation and transmission
of nerve impulses and is manifest by the presence of
normal electroencephalographic (EEG) activity.
Ischemia. Ischemia may be global or focal, as well
as complete or incomplete. Complete global ischemia
occurs with cardiac arrest; incomplete global
ischemia occurs with hypotension or shock. Focal
ischemia involves the occlusion of a single vessel
and is thus incomplete.
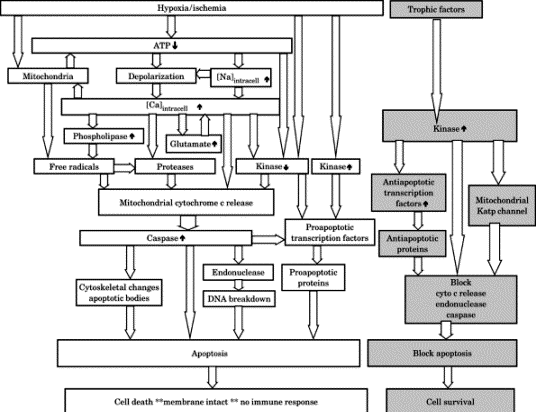
The ischemic cascade. The ischemic cascade occurs
when inadequate cerebral perfusion leads rapidly to
a cascade of pathophysiologic changes involving a
multitude of chemical mediators of neuronal damage.
Decreased availability of oxygen and glucose results
in immediate depletion of ATP, which is required for
all active cellular processes. This depletion occurs
within 2 to 4 minutes of complete ischemia.
Phosphocreatine (PCr) is a source of high energy
phosphate that allows the resynthesis of ATP from
adenosine diphosphate (ADP). Brain PCr levels are
normally three times those of ATP. A decrease in PCr
is one of the earliest harbingers of ischemia.
Lactate levels increase because of anaerobic
metabolism of glucose. Lactic acidosis aggravates
ischemic damage. Lactic acid reduces ferric to
ferrous iron, which in turn promotes free radical
formation followed by lipid peroxidation of cell
membranes. With incomplete ischemia, the persistence
of residual perfusion facilitates increased lactate
production in the presence of ongoing anaerobic
metabolism and is thought to be the mechanism for
increased damage with this type of ischemia. In
contrast, complete ischemia results in complete
cessation of metabolism.
Increased plasma glucose is an independent risk
factor for aggravation of ischemia. The primary
mechanism appears to be increased production of
lactate with intracellular acidosis which
contributes to neuronal necrosis. Hyperglycemia also
prevents the increase in brain adenosine that occurs
with ischemia. Adenosine, a purine nucleotide,
inhibits excitatory amino acid (EAA) release and
promotes cerebrovasodilatation, thus theoretically
attenuating ischemic damage. In addition, there is
some evidence that insulin has neuroprotective
effects independent of its glucose-lowering
properties. Hypoglycemia can also exacerbate
ischemic brain injury. The persistence of
hypoglycemia results in seizure activity and
neuronal injury, particularly to the hippocampus.
Cerebral ischemia increases release of the
EAA
neurotransmitters glutamate and aspartate.
Three receptors for the excitatory neurotransmitters
are currently identified.
(1) N-methyl-D-aspartate (NMDA) receptors are
located in layers three, five, and six of the
cerebral cortex, thalamus, striatum, and Purkinje
fibers, the granule cell layers of the cerebellum,
and the CA1 region of the hippocampus, which is
particularly susceptible to ischemia. NMDA receptors
mediate the influx of sodium (Na) and calcium (Ca)
through membrane channels. Magnesium and the
experimental drug
dizocilipine maleate (MK-801) block the NMDA
receptor site in a noncompetitive fashion.
(2) Quisqualate
(alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic
acid [AMPA]) receptors occur in the deep cortical
layers, thalamus, striatum, molecular layer of the
cerebellum, and pyramidal cell layer and striatum lucidum of the hippocampus. AMPA receptors mediate
the influx of Na.
(3) Kainate receptors, located in the striatum lucidum of the hippocampus, also mediate the influx
of Na.
Glutamate stimulates all three receptors, but
aspartate affects only the NMDA receptor. The
presence of glycine is necessary for activation of
NMDA receptors by glutamate.
Glutamate causes neuronal cell death by two
mechanisms: immediate and delayed. In immediate
neurotoxicity, glutamate activates the NMDA
receptor, leading to Na, chloride (Cl), and water
(H2O) influx, which results in cellular edema,
membrane lysis, and cell death. In delayed
neurotoxicity (24 to 72 hours), the activated NMDA
receptor promotes a cycle of ischemia initiated by
the influx of Ca. This leads to activation of
phospholipases, proteases, and eventually free fatty
acids (FFAs), formation of arachidonic acid and free
radicals, lipid peroxidation, and, ultimately, cell
death.
Increased Ca influx is an early, pivotal event in
the ischemic cascade and is caused by several
mechanisms.
Depletion of ATP results in failure of the
energy-requiring sodium/potassium (Na/K)
ATPase-dependent ion pumps. Na and Cl influx and K
efflux ensue. Influx of H2O and edema occur
secondarily. The resulting membrane depolarization
leads to opening of voltage-sensitive Ca channels
and Ca influx.
Decreased ATP leads to Ca release from endoplasmic
reticulum.
EAA levels increase during ischemia, leading to
stimulation of glutamate receptors and the opening
of NMDA-mediated Ca channels.
Ca extrusion from the cell is an active process that
stops when ATP stores are exhausted.
Numerous ischemic effects of Ca form a common
pathway leading to neuronal cell destruction.
Increased intracellular Ca activates
phospholipase
A1, A2, and C, which leads to the hydrolysis of
membrane phospholipids and the release of FFAs.
Loss of membrane phospholipids also results in
mitochondrial and cell membrane destruction.
Arachidonic acid. The major FFA, arachidonic acid,
is metabolized to prostaglandins, leukotrienes, and
free radicals. Both prostaglandins (via the
cyclooxygenase pathway) and leukotrienes (via
lipoxygenase) cause cerebral edema. Thromboxane A2,
a prostaglandin derived from arachidonic acid with
potent vasoconstrictor and platelet aggregation
properties, potentiates ischemia and has been
implicated in reperfusion injury.
Free radical formation. Superoxide, peroxide, and
hydroxyl radicals cause lipid peroxidation within
neuronal cell membranes. This alters membrane
function and releases toxic by-products (aldehydes,
hydrocarbon gases). These by-products cause edema,
blood-brain barrier disruption, and inflammation.
The superoxide radical itself can create an
inflammatory response with vascular plugging.
 Clinical ischemia
Clinical ischemia
Of all body organs, the brain is the most vulnerable
to ischemia. Loss of consciousness occurs within 15
seconds of cardiac arrest. Brain PCr becomes
negligible within 1 minute. Glucose and ATP stores
are exhausted within 4 to 5 minutes. Critical levels
for cerebral blood flow (CBF), cerebral perfusion
pressure (CPP, the difference between the mean
arterial pressure [MAP] and the intracranial
pressure [ICP]), and the partial pressure of
arterial oxygen (Pao2) have been determined below
which cerebral ischemia occurs (Figure -1) with
characteristic EEG changes.
Critical CBF is 18 to 20 mL/100 g of brain/minute.
The penumbra is a hypoperfused region that may
remain viable depending on timely reperfusion. The
EEG becomes isoelectric at a CBF of 15 mL/100
g/minute. Metabolic failure occurs at a CBF of 10
mL/100 g/minute.
Critical CPP is 50 mm Hg in the normal individual.
Critical Pao2 is 30 to 35 mm Hg in healthy awake
patients.
Reperfusion injury refers to damage that occurs
after the restoration of cerebral perfusion. An
initial phase of hyperperfusion occurs, followed by
a gradual decline in CBF referred to as the
no-reflow phenomenon. Hypoperfusion results from
thromboxane-induced vasoconstriction and platelet
aggregation, impaired red cell deformability, tissue
edema, and the persistence of abnormal Ca levels. In
addition, intracellular acidosis, continued EAA,
neurotransmitter and catecholamine release, and free
radical formation contribute to delayed neuronal
damage. This no-reflow phenomenon can last for up to
24 hours.
|
 |
|
Figure-1. ATP, adenosine
triphosphate. |
III. Clinical cerebral protection
Rationale for treatment. The goal is to maximize the
available oxygen by increasing oxygen supply
(delivery) and decreasing oxygen demand.
Preservation of CBF and the avoidance of hypoxia and
hypoxemia are critical.
Candidates for cerebral protection. Candidates for
cerebral protection include patients who have the
following characteristics:
Patients who have space-occupying lesions such as
tumor, abscess, hematoma, hydrocephalus, and chronic
cystic fluid collections with or without increased
ICP who are scheduled for neurosurgical procedures.
Patients who are scheduled for intracranial vascular
procedures, such as cerebral aneurysm
coiling/clipping and excision of arteriovenous
malformation (AVM) and cavernous angioma, and
extracranial vascular procedures including carotid
endarterectomy (CEA) and superficial temporal artery
to middle cerebral artery (STA-MCA) bypass, which
involve temporary vessel occlusion and the
possibility of focal ischemia.
Patients who are scheduled for the clipping or
coiling of giant or complex basilar artery
aneurysms, which may be facilitated by deep
hypothermic circulatory arrest (DHCA).
Cardiac bypass patients who typically are at risk
from either global ischemia from low-flow states or
focal ischemia from multiple small emboli.
Patients who have had a cardiac arrest with
circulation reestablished within 2 hours.
IV. Clinical therapies
Nonpharmacologic treatment
Hypothermia decreases both metabolic and functional
activities of the brain. Although hypothermia
reduces CMRo2 by roughly 7% for each degree Celsius,
the mechanism is not uniformly linear. The
temperature coefficient (Q10), used to describe the
relationship between temperature and CMRo2, is the
ratio of two CMRo2 values separated by 10°C. For
most biological reactions, the Q10 is approximately
2 (a 50% decrease in CMRo2 for every 10°C decrease
in temperature). Thus, if the normothermic brain
(37°C) can tolerate 5 minutes of complete ischemia,
at 27°C the brain should tolerate 10 minutes of
ischemia. The actual Q10 is 2.2 to 2.4 between 37°C
and 27°C, resulting in a reduction of >50% in CMRo2
at 27°C. Between 27°C and 17°C, the Q10 is
approximately 5. This correlates with the gradual
loss of neuronal function, as demonstrated by an
isoelectric EEG (which occurs between 18°C and
21°C) and the ability of the brain to tolerate more
prolonged ischemia than would be predicted based on
a linear model. Below 17°C, the Q10 is 2.2 to 2.4
again.
However, small decreases in temperature have also
resulted in significant reductions in the damage
from cerebral ischemia. Possible mechanisms of
auxiliary hypothermic protection include decreased
Ca influx, decreased EAA release, blood-brain
barrier preservation, and prevention of lipid
peroxidation. Although mild hypothermia (brain
temperature of 32°C to 35°C) can be
neuroprotective in the animal model, clinical
evidence indicates that it is not beneficial during
aneurysm surgery.
Correlation between esophageal and brain
temperatures should not be assumed. Either tympanic
membrane or nasopharyngeal temperature should
therefore be measured as a more accurate estimate of
brain temperature. Avoidance of hyperthermia is
paramount because above-normal temperatures markedly
increase CMRo2 and exacerbate ischemic damage.
Deep hypothermic circulatory arrest to core
temperatures of 13°C to 21°C might be indicated
for clipping giant or complex basilar artery
aneurysms. Peripheral arterial and large-bore
intravenous catheters are inserted before induction
of anesthesia. After induction, either a central
venous or a pulmonary artery catheter, a second
arterial catheter for phlebotomy, and a lumbar
subarachnoid drain are inserted. Electrophysiologic
monitoring of EEG, somatosensory evoked potentials
(SSEPs), and brain stem auditory evoked potentials
(BAEPs) is begun. The SSEPs persist to 15°C to
18°C and a CBF of 10 to 15 mL/100 g/minute, which
is beyond hypothermic EEG isoelectricity (18°C to
20°C).
Cooling at a rate of 0.2°C/minute is performed by
using a cooling blanket, infusing cold saline, and
decreasing ambient temperature. Barbiturate-induced
burst suppression is initiated and maintained
intraoperatively. Hemodilution to a hematocrit of
28% to 30% is accomplished by phlebotomy; this blood
is reserved in an anticoagulant solution to be
reinfused after termination of bypass for
replacement of essential clotting factors.
The aneurysm is dissected with meticulous attention
to hemostasis before beginning femoral artery-femoral
vein bypass. Heparin, 300 to 400 IU/kg, is
administered, and the
activated clotting time (ACT) is kept between 450
and 480 seconds. Cardiopulmonary bypass is begun
when the patient's temperature is 34°C and
continued until the desired core temperature is
reached. Spontaneous atrial fibrillation may occur
below 30°C, and continuous ventricular fibrillation
frequently occurs below 28°C. To prevent myocardial
ischemic injury, persistent ventricular fibrillation
should be terminated by the administration of
potassium chloride (KCl), 20 to 60 mEq.
Cardioversion with 100 to 250 J may be used to
induce asystole in patients resistant to KCl or in
anephric patients in whom KCl is contraindicated.
MAP should be maintained between 40 to 80 mm Hg
during bypass.
Circulatory arrest occurs between 22°C and 18°C.
The bypass pump is stopped. The duration of
circulatory arrest is limited to aneurysm clip
application time. Bypass is resumed and rewarming
proceeds at 0.2 to 0.5°C/minute. Spontaneous
ventricular fibrillation occurs with rewarming.
Cardioversion (200 to 400 J) is required to restore
sinus rhythm. Extracorporeal bypass is terminated
when the patient's temperature reaches 34°C and
normal sinus rhythm and cardiac output are present.
Inotropic support may be required. The previously
removed whole blood is reinfused to promote normal
coagulation. Heparin is reversed with protamine to
achieve an ACT of 100 to 150 seconds. Complications
of this technique include coagulopathy,
postoperative hemorrhage, metabolic acidosis,
hyperglycemia, myocardial depression, and
dysrhythmias.
Avoidance of hyperglycemia. The current
recommendation is to keep the serum glucose below
150 mg/dL. Serum glucose is monitored frequently,
and hypoglycemia (serum glucose below 60 mg/dL) is
scrupulously avoided.
Avoidance of hypotension, hypoxia, and hypercapnia.
The surgeon may request induced hypertension to
improve CPP during temporary proximal occlusion of
the parent vessel before definitive aneurysmal
clip-ligation. Induced hypotension can be
detrimental in patients at risk for vasospasm.
Hemodilution to a hematocrit of 32% to 34% increases
CBF by decreasing viscosity, thereby improving
oxygen delivery.
Normalization of increased ICP is achieved through
moderate hyperventilation (partial pressure of
arterial carbon dioxide [Paco2] of
25 to 30 mm Hg), head elevation to 30° in the
neutral position, mannitol and/or furosemide
diuresis, cerebrospinal fluid (CSF) drainage via
ventriculostomy, limited fluid restriction, and
barbiturate coma in patients unresponsive to these
techniques.
Correction of acidosis and electrolyte imbalance
including Na and K abnormalities should be prompt.
Pharmacologic treatment
Barbiturates and erythropoietin remain the only
drugs shown to be effective for pharmacologic
cerebral protection against ischemic damage in
humans.
Thiopental, a potent cerebrovasoconstrictor,
decreases CMRo2, CBF, cerebral blood volume (CBV),
and ICP. CO2 reactivity is preserved.
(1) The primary mechanism of protection involves a
reduction in CMRo2 of up to 55% to 60% at which
point the EEG becomes isoelectric. Further reduction
in CMRo2 confers no additional protection.
Thiopental's beneficial effects are thus limited to
preservation of neuronal function.
(2) Thiopental may cause an inverse steal phenomenon
whereby vasoconstriction in normal tissue improves
perfusion of ischemic areas that are unable to
vasoconstrict.
(3) Thiopental is an effective anticonvulsant.
(4) Other possible mechanisms include
gamma-aminobutyric acid (GABA) agonism, free radical
scavenging, membrane stabilization, NMDA antagonism,
Ca channel blockade, and maintenance of protein
synthesis.
(5) Thiopental does not improve outcome in global or
complete ischemia after cardiac arrest.
(6) The thiopental dose in focal ischemia is 3 to 5
mg/kg every 5 to 10 minutes titrated to EEG burst
suppression up to a total of 15 to 20 mg/kg.
Maintenance of cardiovascular stability could
determine the rate of administration.
Pentobarbital's cerebral effects are similar to
those of thiopental. Pentobarbital is longer acting
(t1/2 = 30 hours). The current clinical indication
for pentobarbital is limited to barbiturate coma in
patients who have increased ICP resistant to
standard therapy. A loading dose of 3 to 10 mg/kg
over 0.5 to 3 hours is given, followed by a
maintenance infusion
of 0.5 to 3 mg/kg/hour titrated to EEG burst
suppression. The currently accepted therapeutic
plasma concentration of pentobarbital is 2.5 to 4
mg/dL.
Methohexital, a short-acting barbiturate, can
precipitate seizures in individuals who have
epilepsy. Methohexital is useful for the induction
of anesthesia for brief procedures in which seizure
activity is desired (e.g., electroconvulsive therapy
[ECT] and epilepsy surgery).
Other intravenous anesthetics. Anesthetic drugs that
maintain ATP levels by decreasing cerebral
metabolism while simultaneously preserving CBF and
cardiovascular stability have theoretical potential
for cerebral protection.
Etomidate is a short-acting imidazole compound
which, like barbiturates, causes cerebral
vasoconstriction. Electroencephalographic burst
suppression occurs with higher doses. Most studies
have not shown beneficial effects after cerebral
ischemia. The administration of induction doses of
etomidate has been associated with cerebral
desaturation.
(1) Etomidate reduces CMRo2 (by as much as 50%),
CBF, and ICP while maintaining cardiovascular
stability and CPP. CO2 reactivity is preserved.
(2) Etomidate can cause adrenocortical suppression
for up to 24 hours after a single induction dose
(inhibition of 11 beta-hydroxylase). This may be of
clinical concern when etomidate is used as an
infusion, especially in patients who are not
concomitantly receiving steroids.
(3) Myoclonic activity has been reported with
etomidate, and seizures may occur.
(4) Side effects of etomidate include nausea,
vomiting, and pain on injection.
Propofol (2, 6-diisopropylphenol), a short-acting
induction drug also used to maintain anesthesia, has
a cerebrovascular profile similar to that of
barbiturates. Beneficial effects of propofol after
brain ischemia have not been demonstrated.
(1) Propofol decreases CMRo2, ICP, and CBF (via cerebrovasoconstriction). Hemodynamic depression
decreases CPP more than with barbiturates.
(2) Burst suppression on EEG occurs with larger
doses of propofol.
(3) Propofol may decrease postoperative nausea and
vomiting.
Benzodiazepines, sedative-hypnotic drugs most
commonly used as anesthetic adjuncts,
stimulate the inhibitory neurotransmitter GABA and
decrease CMRo2 and CBF while preserving CO2
reactivity. ICP may be decreased slightly.
Benzodiazepines are potent anticonvulsants. They
also produce amnesia and anxiolysis.
(1) Diazepam is used as an oral premedicant at a
dose of 0.1 to 0.25 mg/kg. Its prolonged t1/2 of 21
to 37 hours limits its use in neurosurgical patients
in whom prompt emergence and postoperative
neurologic assessment are critical. Diazepam remains
an effective treatment for status epilepticus.
(2) Midazolam has a t1/2 of 1 to 4 hours. The
intravenous dose of midazolam for premedication is
0.5 to 2.5 mg up to 0.1 mg/kg. Excessive sedation
and the possibility of hypoventilation-induced
hypercapnia should be avoided in patients at risk
for increased ICP. Midazolam in larger doses may
have beneficial effects after brain ischemia.
(3) Lorazepam is also an effective premedicant in
doses of either 0.5 to 4 mg by mouth or 2 to 4 mg
intravenously (i.v.) or intramuscularly (i.m.). Like
diazepam, its use is limited in neurosurgery by a
t1/2 of 10 to 20 hours.
Opioids produce sedation and analgesia and cause a
reduction in neurotransmitter release while
preserving autoregulation, CO2 reactivity, and
cardiovascular stability. CBF, CMRo2, and ICP are
unchanged or slightly decreased. Delta waves are
seen on EEG; burst suppression does not occur.
(1) Morphine is a potent analgesic with relatively
poor central nervous system (CNS) penetration.
Commonly used for postoperative analgesia in
neurosurgical patients, morphine can cause
hypotension secondary to histamine release.
(2) Meperidine may increase the heart rate because
of its atropine-like structure and effect.
Normeperidine is a metabolite of meperidine that can
cause CNS excitation and seizures.
(3) Fentanyl is 100 times more potent than morphine.
Fentanyl does not cause histamine release, is
shorter acting than morphine, and decreases ICP and
CBV slightly while maintaining CPP.
(4) Sufentanil is more potent than fentanyl and may
increase ICP (via vasodilatation) in patients who
have severe head
trauma. The use of another opioid should be
considered in such instances.
(5) Remifentanil is a very short-acting (t1/2 = 3 to
10 minutes) esterase-metabolized opioid that
compared favorably to fentanyl in reduction of ICP
and CBV and maintenance of CPP in a recent clinical
trial.
Ca channel-blocking drugs should theoretically
provide cerebral protection by vasodilatation and
diminution of the consequences of Ca influx.
(1) Nimodipine decreases vasospasm after aneurysmal
subarachnoid hemorrhage (SAH). Nimodipine may
increase CBF to underperfused areas by
redistribution through an inverse steal effect. The
dose of nimodipine, presently available only in oral
form, is 60 mg every 4 hours for 21 days after SAH.
Hypotension may occur with the administration of
nimodipine.
(2) Nicardipine, available for intravenous
administration, has decreased ischemic damage in
animal studies, but clinical trials have not shown
improved neurologic outcome after ischemia.
Ketamine, a phencyclidine derivative, produces
dissociative anesthesia.
(1) Ketamine markedly increases ICP and CBF (60%)
via cerebrovasodilatation. The CMRo2 is unchanged or
slightly increased. Autoregulation is abolished.
(2) Seizures can occur.
(3) Although it is a noncompetitive NMDA antagonist,
ketamine is not recommended for patients who have
intracranial pathology.
Local anesthetics are commonly used as adjuvants in
neuroanesthesia.
(1) Lidocaine's clinical effects are determined by
the dose. When administered after EEG isoelectricity
induced by pentobarbital, lidocaine may decrease
CMRo2 by an additional 15% to 20%. At clinically
recommended doses (1.5 mg/kg), lidocaine may reduce
ischemic damage. Lidocaine also blunts the
hemodynamic response to intubation by increasing
anesthetic depth. At lower doses, lidocaine
possesses anticonvulsant activity and can be used as
ancillary therapy for status epilepticus. At toxic
doses, lidocaine causes seizures.
Potent inhaled anesthetics
All potent inhaled anesthetics are
cerebrovasodilatators and thereby increase CBF and
ICP to different degrees. This effect can be
attenuated by prior hyperventilation. The volatile
anesthetics also decrease CMRo2 while uncoupling CBF
and CMRo2. Autoregulation is impaired but CO2
reactivity is preserved.
Isoflurane causes the greatest decrease in CMRo2
(40% to 50%) and is the least potent vasodilator.
The EEG becomes isoelectric EEG at 2 minimum
alveolar concentration (MAC) or 2.4%. Isoflurane has
no effect on the production of CSF but does increase
CSF resorption. The critical CBF for isoflurane, the
lowest of all the volatile agents, is 10 mL/100
g/minute. Thus, the use of isoflurane in patients
undergoing CEA may have advantages. Isoflurane may
also offer protection after brain ischemia. Studies
of isoflurane in animal models of ischemia and
hypoxemia have shown some limited protection from
isoflurane. Preconditioning with isoflurane seems to
confer tolerance to ischemia and some
neuroprotection. In vitro studies have also shown
improved recovery after ischemia and a reduction in
cell death through the postischemic activation of
ATP-regulated K channels and protein kinases.
The cerebral effects of sevoflurane are similar to
those of isoflurane; both cause a slight increase in
CBF and ICP and a decrease in CMRo2. Nephrotoxic
inorganic fluoride may accumulate when patients
receive sevoflurane for prolonged periods of time.
Induction and emergence are rapid. Sevoflurane may
offer protection after brain ischemia through
preconditioning. Preconditioning with sevoflurane
and subsequent cerebral protection have been
demonstrated during incomplete ischemia in vitro.
Improved recovery in CA1 pyramidal cells in rats has
occurred at clinical concentrations known to be
useful in humans.
Desflurane is similar to isoflurane in its
cerebrovascular profile, but ICP might increase
despite normocapnia with desflurane compared to
isoflurane. Induction and emergence with desflurane
are rapid. Desflurane may also be protective after
brain ischemia. Studies after hypoxia and after
incomplete cerebral ischemia in rats have shown
cerebral protective effects.
Nitrous oxide, a cerebrovasodilatator, increases
CBF, CMRo2, and ICP. The increase in CBF
is attenuated by barbiturates, opioids, and
hypocapnia. Nitrous oxide is 32 times more soluble
in blood than nitrogen and is thus capable of
diffusing into air-containing body cavities with
extreme rapidity. Therefore, nitrous oxide is
avoided in the presence of pneumocephalus and in any
surgical procedure within 2 weeks of a craniotomy in
which nitrous oxide was used. Nitrous oxide is also
discontinued immediately if air embolism is
suspected and may increase neurologic deficits after
brain injury.
Anticonvulsant drugs are indicated in patients at
risk for seizure activity including individuals who
have epilepsy, head trauma, or craniotomy, and their
administration is continued into the postoperative
period. Seizure activity exacerbates the effects of
ischemia through activation of anaerobic metabolic
pathways. CBF, CMRo2, and intracellular Ca increase
during seizures, and EAA neurotransmitters,
including glutamate, are released.
Once seizure activity occurs, the patient's airway
is immediately secured and adequate ventilation is
ensured to prevent hypoxemia and hypercapnia.
Avoidance of hypotension is essential.
Anticonvulsant therapy is administered promptly:
Thiopental, 25 to 100 mg i.v.
Diazepam, 2 to 20 mg i.v.
Midazolam, 1 to 5 mg i.v.
Fosphenytoin, 15 to 20 mg phenytoin equivalents
(PE)/kg, or phenytoin, 15 mg/kg, may be administered
to prevent further seizure activity once the acute
episode has been terminated. Fosphenytoin, 75 mg, is
equivalent to phenytoin, 50 mg, and has the
advantage of increased speed of administration (up
to 150 mg PE/minute). Phenytoin is limited to 50
mg/minute because it may induce hypotension. The
loading dose of fosphenytoin can be given in 5 to 7
minutes, whereas the equivalent dose of phenytoin
would require 15 to 20 minutes.
V. Cerebral preconditioning and neurogenesis
Models of cerebral ischemia in animals have shown
that the induction of endogenous proteins of repair
and genes that code for them can set the stage for
cerebral preconditioning that may protect the brain
during subsequent ischemia. Prodromal transient
ischemic attacks (TIAs) may protect the brain during
subsequent ischemic strokes. Recent evidence
indicates that neurogenesis and diaschisis occur
after injury. Diaschisis is a reduction in blood
flow and metabolism in an area distant from the site
of focal damage. It may represent a process of
structural reorganization after injury. Apoptosis,
or programmed cell death, may also be part of the
process of structural reorganization after injury.
Ischemia stimulates neurogenesis and new neurons
migrate to the site of tissue injury and contribute
to functional recovery. Activated neural stem cells
contribute to stroke-induced neurogenesis and the
migration of neuroblasts toward the infarct boundary
in adult rats. Therapy for stroke in rats with a
nitric oxide (NO) donor and human bone marrow
stromal cells enhances angiogenesis and neurogenesis
subsequent to middle cerebral artery occlusion.
VI. Cerebral resuscitation
Patients who require resuscitation from cerebral
ischemia include the following:
Intensive care unit (ICU) patients who have
traumatic but nonoperative brain injury such as
diffuse axonal injury (DAI) with increased ICP and
cerebral edema who may be candidates for barbiturate
coma.
Patients who have Reye's syndrome and cerebral edema
with increased ICP.
Near-drowning victims who have anoxic
encephalopathy, cerebral edema, and intracranial
hypertension who are treated like Reye's syndrome
patients.
Patients who have nonhemorrhagic stroke who may be
candidates for fibrinolytic therapy with tissue
plasminogen activator (TPA).
Anesthesiologists may encounter these patients when
they are consulted about the management of cerebral
edema and increased ICP or the induction and
maintenance of barbiturate coma. These patients may
also require sedation, analgesia, and neuromuscular
blockade.
VII. Experimental modalities
NMDA receptor antagonists
were developed to prevent
neuronal damage from the excessive accumulation of
the excitatory neurotransmitter glutamate. The NMDA
receptor antagonists have not conferred consistently
reproducible neuroprotection in experimental studies
and may worsen injury. One of the difficulties has
been the development of drugs that effectively
penetrate the blood-brain barrier.
Dizocilpine maleate (MK-801) is a noncompetitive
NMDA receptor antagonist whose beneficial effects in
laboratory experiments may be partially attributable
to drug-induced hypothermia. Dizocilpine is not
approved for use in humans and does not appear to be
promising.
Magnesium, a noncompetitive NMDA antagonist, binds
within the ion channel, preventing ion flux, and may
be helpful after brain injury.
Glycine binding site antagonism with HA-966 and
7-chlorokynurenic acid is still in the
investigational stage but shows promise.
AMPA receptor antagonism with 2,
3-dihydroxy-6-nitro-7-sulfamoylbenzo (f)quinazoline
(NBQX) has proved beneficial when given after the
ischemic insult in experimental models.
Sodium channel-blocking drugs such as riluzole may
reduce glutamate release during ischemia.
Lamotrigine, an anticonvulsant with Na
channel-blocking activity, is known to reduce
glutamate release and ischemic damage. Further
studies are warranted.
Tirilazad, a lipid-soluble 21-aminosteroid, crosses
the blood-brain barrier and acts as a lipid
antioxidant, inhibiting free radical formation and
lipid peroxidation. Studies indicate protection only
when tirilazad is administered before an ischemic
event.
Free radical scavengers. Superoxide dismutase (SOD),
deferoxamine, vitamin E, mannitol, and
glucocorticoids all possess free radical scavenging
activity. The utility of SOD has been limited by its
short t1/2 (8 minutes) and poor blood-brain barrier
penetration. While glucocorticoids have
membrane-stabilizing properties and decrease
cerebral edema from brain tumors, they have not been
shown to improve outcome in cerebral ischemia. The
clinical usefulness of free radical scavengers is
still under investigation.
Modification of arachidonic acid synthesis.
Ischemia-induced excess of the vasoconstrictor
thromboxane relative to the vasodilator prostacyclin
(PGI2) has led to the development of thromboxane
synthetase inhibitors and PGI2 synthetase
stimulation to prevent the formation of excessive
thromboxane.
Dexmedetomidine, an alpha2 agonist, decreases
central sympathetic activity by decreasing plasma norepinephrine release. Dexmedetomidine has been
found to be neuroprotective in a model of focal
ischemia, perhaps because excess catecholamine
levels correlate with increased neuronal ischemic
damage. Dexmedetomidine also decreases the MAC for
halothane and isoflurane and decreases CBF without
significantly altering CMRo2.
NO is a free radical with complex neuronal activity.
Nitric oxide synthase (NOS) catalyzes the formation
of NO from the amino acid L-arginine, which itself
decreases neuronal damage in experimentally induced
focal ischemia. Three forms of NOS have been
discovered:
Neuronal NOS (nNOS) enhances glutamate release and
NMDA-mediated neurotoxicity. Selective nNOS
inhibition has been shown to be neuroprotective.
Immunologic NOS (iNOS) is not detectable in healthy
tissue. Induction of iNOS causes delayed neuronal
cell death and can exacerbate glutamate
excitotoxicity. Inhibition of iNOS by aminoguanidine
reduces ischemic damage in experimental models.
Stimulation of endothelial NOS (eNOS) by an
ischemia-induced increase in intracellular Ca
improves CBF by dilatation of cerebral blood vessels
and has been shown to reduce ischemic damage in a
rodent model.
Erythropoietin (EPO) is a substance produced in the
brain after hypoxic or ischemic insults. Primarily
elaborated in the adult mammalian astrocytes in the
ischemic penumbra, EPO stimulates neurogenesis,
angiogenesis, and production of the proteins of
repair, diminishes neuronal excitotoxicity, reduces
inflammation, and inhibits neuronal apoptosis. It
has been used in humans for cerebral preconditioning
in patients after ischemic stroke. EPO may be more
effective, however, as a prophylactic protectant
when given preoperatively. Nonhematopoietic analogs
of EPO, such as asialoEPO, have been developed and
are showing equivalent potency as neuroprotectants
in the laboratory. These analogs do not increase the
hematocrit and thus do not exacerbate the ischemia
injury through an increase in blood viscosity.
Other experimental modalities. Experimental results
with preoperative hyperbaric oxygen, normobaric 100%
oxygen exposure, electroconvulsive shock, and the
potassium channel-opening drug, diazoxide, have
shown that all of these modalities can be used to
accomplish cerebral preconditioning.
Anesthesia duration and depth.
Minimizing the
duration of the time during which the patient is
deeply anesthetized may provide cerebral protection
by preventing neuronal apoptosis. A growing body of
evidence indicates that cumulative deep anesthesia
time is an independent predictor of increased
postoperative mortality in adult patients.
|
