|
| Postoperative
Complications |
Respiratory Care|
Cardiovascular Therapy|
Fluid Management| Nutrition in Critical Patients
|Traumatic Brain Injury-Stroke-Brain Death
|

Specific considerations of
respiratory pathology and care in the neurosurgical
intensive care unit (NICU) are discussed. The
development of pulmonary complications contributes
significantly to mortality in critically ill
neurosurgical patients and worsens neurologic
outcome. Prompt recognition and treatment of
pulmonary disorders are therefore important in the
management of neurosurgical patients.
I. Respiratory
disorders
 Clinical physiology of
oxygen transfer. Arterial hypoxemia
commonly occurs in patients after central nervous
system (CNS) injury, adversely affecting neurologic
outcome, especially in patients who have sustained
head trauma. Arterial hypoxemia (PaO2 <50
mm Hg) causes cerebral vasodilatation and increases
in cerebral blood flow and cerebral blood volume,
which could have a detrimental impact on
intracranial pressure (ICP). The major causes of
arterial hypoxemia are (a) a decrease in alveolar
oxygen tension (Pao2) owing either to
hypoventilation or to decreased inspired oxygen
tension (Pio2) or (b) an increase in
alveolar-arterial oxygen tension difference (P[A-a]O2).
Inspired gas is mixed with gas in the functional
residual capacity (FRC) to make up alveolar gas.
Mixed venous blood equilibrates with alveolar gas
and is distributed to the systemic circulation as
mixed arterial blood.
Clinical physiology of
oxygen transfer. Arterial hypoxemia
commonly occurs in patients after central nervous
system (CNS) injury, adversely affecting neurologic
outcome, especially in patients who have sustained
head trauma. Arterial hypoxemia (PaO2 <50
mm Hg) causes cerebral vasodilatation and increases
in cerebral blood flow and cerebral blood volume,
which could have a detrimental impact on
intracranial pressure (ICP). The major causes of
arterial hypoxemia are (a) a decrease in alveolar
oxygen tension (Pao2) owing either to
hypoventilation or to decreased inspired oxygen
tension (Pio2) or (b) an increase in
alveolar-arterial oxygen tension difference (P[A-a]O2).
Inspired gas is mixed with gas in the functional
residual capacity (FRC) to make up alveolar gas.
Mixed venous blood equilibrates with alveolar gas
and is distributed to the systemic circulation as
mixed arterial blood.
Causes of decreased Pao2.
Pao2, the major driving factor for
arterial oxygenation, may be reduced because of
either a decrease in Pio2 or diminished
alveolar ventilation from respiratory depression,
increased dead space, or mechanical impairment. Pao2
is determined by the barometric pressure (PB), the
fraction of inspired oxygen (Fio2), and
the ratio of oxygen consumption ([V with dot above]O2)
to alveolar ventilation ([V with dot above]A):
PAO2 = PB x ( FIO2
- VO2 /VA).
As far as [V with dot above]A is directly
proportional to carbon dioxide production
([V with dot above]CO2), [V with dot
above]A = [V with dot above]CO2/PACO2
× (k), and PACO2 ~ PaCO2,
Equation .1 can be transformed into the alveolar gas
equation .2:
PAO2 = FIO2(PB-47)-
(PaCO2/Rq).
where RQ = respiratory quotient, RQ
= [V with dot above]CO2/[V with dot
above]O2 (0.8 in resting state), and 47 =
saturated water vapor pressure at the PB in mm Hg.
 Respiratory depression
(hypoventilation). Respiratory depression can
be either central (from CNS disease, narcotics, or
sedatives) or peripheral (from neuromuscular
disease, muscle relaxants).
Respiratory depression
(hypoventilation). Respiratory depression can
be either central (from CNS disease, narcotics, or
sedatives) or peripheral (from neuromuscular
disease, muscle relaxants).
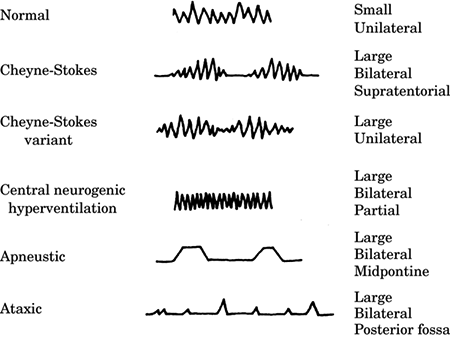
1. Traumatic brain injury (TBI). Variations in both the
depth and rate of spontaneous respirations occur in
60% of patients who have CNS injury. Five
respiratory patterns are observed in patients who
have head injuries and brain tumors (Fig.-1).
Pathologic hypoventilation and hyperventilation lead
to hypoxemia, pH disturbances, and electrolyte
imbalance.
|
Figure -1. Respiratory patterns associated with
closed head injury. |
|
 |
|
Normal breathing in an awake patient with a small,
unilateral lesion. Cheyne-Stokes ventilation is a
periodic breathing pattern in which hyperpnea
alternates with apnea. The hyperpneic phase lasts
longer than the apneic phase. The Cheyne-Stokes
variant has a shorter apneic phase and is associated
with large unilateral injuries. Central neurogenic
hyperventilation is a sustained, regular, rapid,
fairly deep hyperpnea. It occurs with a large
pontine lesion, systemic hypoxia, or metabolic
acidosis. It is often associated with brain stem
tumors, including lymphomas and astrocytomas, and
metastatic disease. Apneustic breathing occurs with
large bilateral midpontine lesions. There is a brief
2- to 3-second pause at the end of inspiration,
alternating irregularly with expiratory pauses. It
has a very poor prognosis. Ataxic respiration is a
pattern of irregular breathing in which shallow and
deep breaths alternate randomly with irregular
pauses. It is associated with medullary compression
and has a very poor prognosis. The irregularity
distinguishes it from Cheyne-Stokes ventilation. |
2. Spinal cord injury (SCI). High cervical spinal cord
lesions impair ventilation because of either partial
or complete loss of innervation of the diaphragm
(C3-5), with loss of the ability to cough. The
diaphragm is spared when injury is below C6-7.
However, injury at the thoracic level results in the
loss of intercostal and abdominal muscle function,
which reduces FRC and creates paradoxical
ventilation (chest retraction on inspiration and
expansion during expiration), loss of cough, and
reduced ability to handle secretions. All lung
volumes, except residual volume, are markedly
decreased. With a C5-6 lesion, ventilation may be
inadequate and forced vital capacity (FVC) decreased
to 30% of predicted capacity. Improvement of FVC
occurs with time because of the strengthening of the
accessory muscles (clavicular portion of the
pectoralis major and sternocleidomastoid),
development of spasticity (which stops the
paradoxical chest wall motion), improvement of
diaphragmatic function, and decreased usage of
narcotics and sedatives. At 5 months after injury,
FVC reaches 60% of predicted values.
3. Associated airway injuries in multiple trauma
patients including tracheobronchial fistula,
tracheobronchial disruption, and rupture of
diaphragm cause acute hypoventilation and profound
hypoxemia.
Increased airway dead space. Dead space increases
with drug administration (such as atropine and
nitroprusside for deliberate hypotension),
mechanical ventilation with positive end-expiratory
pressure (PEEP), hypovolemia, hypotension,
hypocapnia, and cardiopulmonary disease.
Mechanical impairment. Airway obstruction from
oropharyngeal soft tissue, uncleared secretions,
hemoptysis, foreign bodies, bleeding, or
bronchospasm can cause mechanical impairment of
effective alveolar ventilation. The restriction of
free chest movement from surgical positioning,
surgical retraction of the chest and abdomen,
traumatic rupture of the diaphragm, pneumothorax,
hemothorax, flail chest and restrictive lung disease
can reduce alveolar ventilation significantly.
Proper recognition and treatment of traumatic chest
injuries are crucial to the successful management of
patients after traumatic injury.
Causes of increased P(A-a)O2. The P(A-a)O2 may be
increased as the result of both pulmonary and extrapulmonary factors. Pulmonary disorders can
increase P(A-a)O2 because of a (a) shunt, (b) [V
with dot above]A/[Q with dot above] mismatch, and
(c) limitation of diffusion. Most pulmonary
disorders increase the [V with dot above]A/[Q with
dot above] mismatch and shunt but seldom affect
diffusion capacity. Decreases in cardiac output and
either a respiratory or metabolic alkalosis are
important extrapulmonary factors that increase
P(A-a)O2 in the presence of lung disease.
Extrapulmonary factors. Two extrapulmonary factors
that commonly contribute to hypoxemia in
neurosurgical patients who have preexisting lung
disease are (a) hyperventilation to reduce ICP and
(b) excessive depletion of intravascular volume from
fluid restriction and the administration of
diuretics.
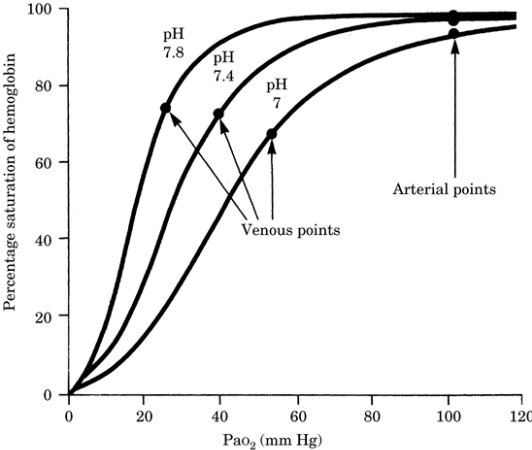
1. Respiratory alkalosis. Hyperventilation to induce
hypocapnia results in a decrease in venous return
and cardiac output. Hypocapnia increases pulmonary
shunt, especially in the presence of lung disease.
The mechanism is unclear but might be related to hypocapnic bronchoconstriction and inhibition of
hypoxic pulmonary vasoconstriction. In addition, the
oxyhemoglobin dissociation curve shifts to the left,
resulting in a lower Pa O2 for any given oxygen
saturation (Fig.-2). This is especially
noticeable when the Pa O2 is equal to or <70 mm Hg.
| Figure -2. Shift in the oxyhemoglobin dissociation
curve with change in blood pH. |
 |
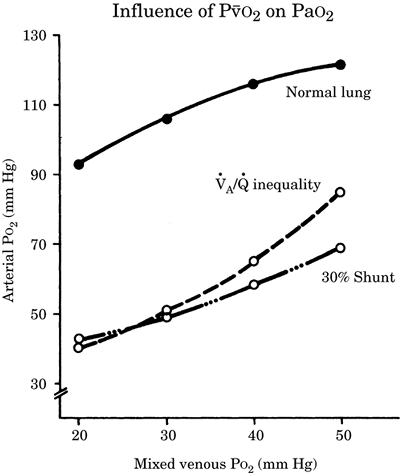
2. Hypovolemia and decrease in P[ v with bar above]O2.
Pa O2 is lower when the mixed venous oxygen tension
(P[v with bar above]O2) is decreased, as can occur
with decreased cardiac output, increased oxygen
consumption, or severe anemia with increased oxygen
extraction. The effect of P[v with bar above]O2 can
be quite significant, especially when there are
areas of low [V with dot above]A/[Q with dot above]
and shunt (Fig.-3). For instance, in the presence
of [V with dot above]A/[Q with dot above] mismatch,
PaO2 = 80 mm Hg when P[v with bar above]O2 is 50 mm
Hg. If P[v with bar above]O2 is decreased to 20 mm
Hg from a decrease in cardiac output, then Pa O2
decreases to 40 mm Hg without any change in the
degree of [V with dot above]A/[Q with dot above]
inequality or shunt. Therefore, low cardiac output
from hypovolemia or myocardial depression increases
P(A-a)O2 and might result in hypoxemia, especially
in the presence of pulmonary disease.
Pulmonary factors that increase P(A-a)O2 in
critically ill neurosurgical patients consist of
neurogenic pulmonary edema (NPE), [V with dot
above]A/[Q with dot above] mismatch, and
abnormalities of lung parenchyma.
|
Figure-3. Influence of mixed venous oxygen
tension on arterial oxygen tension. |
 |
a. Neurogenic etiologies of increased P(A-a)O2
(1) Reduced FRC. The FRC is often reduced after TBI
and SCI at the cervical and thoracic levels. Head
trauma patients might exhibit a reduction in FRC and
pulmonary compliance and an increase in pulmonary
shunting without evidence of pulmonary disease on
the chest radiograph.
(2) NPE. Classic NPE develops in severe CNS injury,
such as TBI, SCI, and subarachnoid hemorrhage (SAH).
Of immediate onset, NPE becomes clinically
recognizable 2 to 12 hours after injury. NPE is
generally of short duration and resolves within
hours to days. The pathogenesis of NPE is complex
and related to circulatory hyperactivity owing to
catecholamine release, particularly norepinephrine,
which causes an increase in the permeability of the
alveolar capillary barrier.
(3) Neurogenic alterations in [v with bar above]A/[Q
with dot above] matching. Hypoxemia and increased
pulmonary shunting have been observed in animals and
patients who have CNS disorders in the absence of
pulmonary disease. The etiology of these changes in
[V with dot above]A/[Q with dot above] regulatory
mechanisms is multifactorial. Release of
inflammatory and anti-inflammatory mediators in
patients after TBI may cause acute lung injury
(ALI). Brain thromboplastin is released into the
blood with TBI, causing deposition of fibrin
microemboli and platelet aggregates in the pulmonary
capillaries. Neutrophil accumulation and the release
of vasoactive substances might also cause local [V
with dot above]A/[Q with dot above] abnormalities.
Patients who have an increase in fibrin spit
products are also more likely to develop respiratory
failure.
Pulmonary etiologies of increased P(A-a)O2
(1) Gastric aspiration. Aspiration of gastric
contents occurs frequently in comatose neurosurgical
patients and in trauma patients because of
intoxication from alcohol and street drugs and
abdominal injury. Aspiration of large volumes of
fluid with a low pH or particulate matter results in
aspiration pneumonitis and ALI. The aspiration of
large foreign bodies, such as gravel, gum, or teeth,
might also cause bronchial obstruction, atelectasis,
and hypoxemia, and potentially may lead to
necrotizing pneumonia. Treatment of aspiration is
primarily supportive. Antibiotics are not indicated.
(2) Atelectasis and
lung consolidation are common in the neurosurgical
patient. Central respiratory depression, cervical
and thoracic spine injury, hypoventilation from
pain, altered levels of consciousness, bronchial
obstruction from mucous plugs or aspiration of
particulate matter, and compression of adjacent lung
by traumatic pneumothorax, hemothorax, or pleural
effusion all contribute to the formation of
atelectasis. Prevention and treatment of lung
collapse through hyperventilation, recruitment
maneuvers (PEEP, inverse-ratio ventilation where
inspiration is longer than expiration, "sigh" breaths), and
aggressive physiotherapy can prevent the development
of lung infection. Because SCI patients are not able
to clear secretions adequately, therapeutic
bronchoscopy, bed rotation, and turning the patient
into the prone position can help clear secretions.
(3) Nosocomial pneumonia (NP)/ventilator-associated
pneumonia (VAP). VAP is an NP that develops in
patients 48 hours or more after endotracheal
intubation. NP is the second most common (after
urinary tract infection) and the most serious
infection in hospitalized patients. VAP occurs in 5%
to 30% of intensive care unit (ICU) patients and is
responsible for 30% to 40% of all ICU-acquired
infections. NP/VAP develops in about half of
critically ill neurosurgical patients who have
isolated TBI and SCI and in nearly two-thirds of
patients after cervical SCI. Pulmonary
complications, particularly pneumonia, represent an
independent risk factor for death in SCI.
(a) Risk factors for NP/VAP are presented in Table-1.
(b) Early onset NP/VAP and late-onset VAP are
distinguished by their respective times of onset and
pathogens. In the general ICU population, early
onset NP/VAP develops within the first 5 days of
hospitalization while late-onset VAP develops after
5 days. Early onset NP/VAP is characterized by
community-acquired microorganisms including
gram-positive cocci (Staphylococcus aureus,
Haemophilus influenzae, Streptococcus pneumoniae),
Enterobacteriaceae, and anaerobes. In late-onset VAP,
the causative pathogens are hospital-acquired
(nosocomial), highly virulent, antibiotic-resistant
microorganisms (e.g., Pseudomonas aeruginosa,
Acinetobacter baumannii, Klebsiella, methicillin-resistant
Staphylococcus aureus [MRSA], vancomycin-resistant
Enterococcus). The extreme virulence and the
antibiotic resistance of the flora are responsible
for the high mortality rate of 40% to 60%, which can
go as high as 80% in late-onset VAPs.
Community-acquired organisms predominate as the
causative agents of pneumonia in critically ill TBI
patients in the first 10 days after admission. With
comatose patients, VAP develops most frequently
between hospital days 4 and 7.
|
Table-1. Diagnostic criteria for nosocomial
pneumonia/ventilator-associated pneumonia |
|
Clinical Criteria |
Bacteriological Criteria |
|
I. "Classic" clinical criteria (sensitivity and
specificity 50%-60%)
A.
Change in chest x-ray: new and persistent pulmonary
infiltrates, or new pleural effusion, and
B.
Two or more findings from the following:
1.
Fever >38�C or hypothermia <36�C
2.
White blood cell count (mm3)
≥12,000 or
≤5,000
3.
Purulent tracheobronchial secretions
4.
Impaired oxygenation |
I.
Noninvasive techniques
A.
Endotracheal tube aspirates
1.
Qualitative cultures (sensitivity 90%, specificity
<50%)
2.
Quantitative culture ≥ 105-106 cfu/mL
B.
Mini-BAL with aspirates from lower respiratory tract
with blind protected telescopic catheters
(quantitative cultures) |
I.
Invasive bronchoscopic techniques with quantitative
cultures (sensitivity and specificity 70%-90%)
A.
BAL: ≥104 cfu/mL
B.
Protected specimen brushing: ≥103 cfu/mL
C.
Gram stain: ≥5% infected leukocytes (containing
intracellular microorganisms) is optional |
| II.
Clinical Pulmonary Infection Score
≥6 (includes
scoring system with points for temperature, white
blood cell count, tracheal secretions, oxygenation,
pulmonary radiography, tracheal aspirate) |
|
|
| BAL, bronchoalveolar lavage; cfu/mL, colony-forming
units per milliliter. |
(c) Pathogenesis of late-onset NP/VAP in terms of
sources of infection includes major aspirations,
nasal colonization by S. aureus, and microaspiration
of colonized oral, upper airway, and gastric
secretions, all of which can lead to the development
of microabscesses in the lung. Hypoventilation with
resultant insufficient alveolar aeration and
consequent alveolar collapse and consolidation
hastens the onset of infection.
(d) Diagnosis (Table-2).
Unfortunately, there is no "gold standard" for diagnosis of VAP. Along
with clinical criteria, early bacteriological
detection of causative microorganisms is important
for appropriate antibiotic treatment. If not
supported by bacteriological results, empiric
antibiotic treatment is often inadequate. The use of
invasive bronchoscopic techniques (bronchoalveolar
lavage and protective specimen brushing) with
quantitative cultures of the specimen allows precise
adjustment of antibiotic therapy. However,
finalizing quantitative cultures takes 2 to 3 days,
which inevitably leads to delay in instituting
appropriate treatment.
|
Table-2. Risk factors for nosocomial
pneumonia/ventilator-associated pneumonia |
|
Patient Related
|
Intervention Related
|
| Aspiration |
Endotracheal intubation
|
| Bacterial
colonization of nasopharynx |
Mechanical
ventilation for >48 hours |
| Coma, decreased level of consciousness
|
| Atelectasis |
Barbiturate use |
| Older age |
Traumatic brain injury |
| >30 in early onset
pneumonia |
Histamine type 2
blocking drugs, antacids |
| >60 in late onset
pneumonia |
Muscle relaxants |
| Serum albumin <2.2 |
Excess sedation |
| Adult respiratory
distress syndrome |
Supine position
|
| Chronic obstructive
pulmonary disease |
Frequent
ventilator circuit changes |
| Multiorgan
dysfunction syndrome |
Reintubation/unplanned extubation |
| High gastric pH |
Intracranial pressure monitoring |
| Sinusitis |
Nasogastric tube/enteral nutrition |
|
Uremia |
Parenteral nutrition |
| Patient transport |
| Length of stay in intensive care unita |
| aControversial in long-term intensive care unit
survivors. |
(e) Choice of antibiotic treatment
depends on onset
of NP/VAP, risk factors, and the severity of the
illness. In general, in early onset NP/VAP,
monotherapy with amoxyclavulanate, a
second-generation or nonpseudomonal third-generation
cephalosporin, or carbapenem for 5 to 7 days is
effective in early onset NP/VAP. Late VAP requires
broad coverage of resistant pathogens, and empiric
combination therapy with two or three antibiotics
should be initiated with an antipseudomonal
cephalosporin or carbapenem with amikacin or
quinolone. For MRSA coverage, linezolid or
vancomycin should be added, although linezolid is
more effective than vancomycin. After quantitative
cultures have been finalized, empiric antibiotic
coverage should be adjusted to the particular
pathogen and continued for at least 7 to 21 days,
depending on the microorganism and the severity of
illness.
(f) Preventive strategies for VAP are directed at
preventing aerodigestive tract colonization,
aspiration, and lung consolidation. Routine hand
washing with either water and soap or alcohol gel is
the most effective strategy for reducing
colonization of nosocomial infections. Other
modalities include restriction of antibiotic usage,
attention to oral hygiene, isolation of patients who
have antibiotic-resistant pathogens, and selective
digestive decontamination. Semirecumbent
positioning, subglottic drainage, prevention of
unexpected extubation, and avoidance of nasal
intubation, gastric overdistention, and oversedation
can reduce the chance of aspiration.
(4) Acute lung injury (ALI)/Adult respiratory
distress syndrome (ARDS)
(a) Definition. ALI/ARDS is a clinical syndrome of
diffuse lung injury that results in noncardiogenic,
high-permeability pulmonary edema. ALI/ARDS consists
of the following: (a) acute onset, (b) bilateral
pulmonary infiltrates on the chest radiograph, (c)
noncardiogenic origin of edema: pulmonary capillary
occlusion pressure of <18 mm Hg, (d) hypoxemia, as
defined by a ratio of the partial pressure of
arterial oxygen to the fraction of inspired oxygen
(Pa O2/FIO2) that is
≤300 in ALI, and
≤200 in ARDS. The incidence of ALI/ARDS in North America and
Europe varies from 1.5 to 75 cases/100,000
population. ALI/ARDS is a severe disease with a
significant mortality rate of 22% to 32% for ALI and
approximately 40% to 70% for ARDS. One-third of ALI
cases progress to ARDS in the first week,
particularly in trauma patients. One-third of
patients who have isolated TBI develop ALI/ARDS;
this significantly increases mortality and worsens
neurologic outcome. The major cause of death in
patients with ALI/ARDS is sepsis and multiorgan
failure. Only approximately 10% to 20% of patients
dying from ARDS die from hypoxemia per se.
(b) Risk factors (Table-3). Primary ARDS is
caused by direct lung injury and primarily pneumonia
and has a higher mortality rate than that of
secondary ARDS, which is caused by indirect lung
injury.
(c) Pathogenesis and clinical course. ALI/ARDS
usually develops within the first 72 hours of the
initiating event with 60% to 70% of cases occurring
within the first 24 hours. The initial step in the
pathogenesis of ALI/ARDS is endothelial cell
activation in the pulmonary microvasculature by
various insulting agents such as microbial products,
different cytokines, and histamine, which leads to
microcirculatory distress, microcoagulation,
neutrophil activation and imbalance between
proinflammatory and anti-inflammatory mediators,
impairment of the alveolar-capillary barrier, and
influx of protein-rich fluid into the alveoli. The
alveolar epithelium is damaged, resulting in the
disruption of the epithelial ion and fluid transport
system, impairment of surfactant production, and
loss of the epithelial barrier. Disorganized
epithelial repair leads to fibrosis. The degree of
epithelial injury correlates with the severity of
illness and outcome. Significant abnormalities of
pulmonary vascular permeability usually persist for
1 week after the onset of ARDS. The acute phase may
either resolve in the first week or progress to
fibrosis with persistent hypoxemia. Patients who do
not improve in the first week have a poor prognosis.
In late phases, pulmonary hypertension can develop
from elevated pulmonary vascular resistance,
primarily from arterio-capillary obstruction, rather
than pulmonary vasoconstriction. Pulmonary
compliance decreases. The chest computed tomographic
(CT) scan often shows diffuse interstitial
thickening and honeycombing. In survivors of ARDS,
muscle wasting and weakness are the most prominent
extrapulmonary conditions that persist for a year
after recovery.
|
Table-3. Risk factors for adult respiratory
distress syndrome |
|
Primary ARDS: Direct Lung
Injury |
Secondary ARDS: Indirect
Lung Injury |
| Pneumonia |
Sepsis |
| Aspiration |
Trauma with shock |
| Pulmonary contusion
|
Blood transfusions |
| Fat emboli |
Subarachnoid hemorrhage |
|
Near drowning |
Traumatic brain injury |
| Acute pancreatitis |
| ARDS, adult respiratory distress syndrome. |
Ventilator-induced lung injury (VILI)/ventilator-associated
lung injury is an inflammatory syndrome of diffuse
alveolar damage that worsens the outcome in patients
who have ALI/ARDS. Several mechanisms are suggested
for the pathophysiology of VILI: oxygen toxicity,
alveolar overdistension from either high tidal
volumes (volutrauma) or high airway pressures (barotrauma),
and repeated alveolar opening and collapse during
the ventilatory cycle. Because ALI/ARDS is a
heterogenous lung disease, positive-pressure
ventilation causes uneven distribution of volume and
alveolar pressure to the extent that even moderate
tidal volumes can overstretch healthy alveoli,
leading to inflammatory responses and exacerbation
of inflammatory processes. PEEP diminishes VILI by
recruiting alveoli and preventing alveolar collapse.
Treatment. Lung protective strategies for mechanical
ventilation have contributed to the decline in
mortality along with the prevention and treatment of
nosocomial infection such as pneumonia and sepsis,
prophylaxis against upper gastrointestinal bleeding,
and prevention of thromboembolism.
Lung-protective ventilation is the cornerstone of
ALI/ARDS management. Positive-pressure ventilation
with a tidal volume of 6 mL/kg of ideal body weight
and plateau pressure <30 cm H2O significantly
decreased mortality compared to conventional
ventilation. PEEP should be adjusted to allow for an
Fio2 of <0.6 to prevent oxygen toxicity. The
oxygenation goal is either a Pa O2 of >60 mm Hg or
an oxygen saturation of 90% or better.
Permissive hypercarbia. Lung-protective ventilation
can inevitably lead to hypoventilation. Hypercarbia
with a Paco2 of 60 mm Hg to 100 mm Hg and a pH of
7.25 can be safely tolerated. However, because CO2
is a potent cerebral vasodilator, permissive
hypercarbia is contraindicated in neurosurgical
patients who have intracranial hypertension.
Prone position. Turning the patient from the supine
to the prone position improves oxygenation through
favorable redistribution of ventilation-perfusion
matching by recruitment of collapsed alveoli. In
patients who have SAH and normal ICP, the prone
position enhances brain tissue oxygenation but can
increase ICP slightly. Risks of the prone position
include accidental extubation, loss of intravenous
access, hemodynamic instability, coughing, and late
complications such as edema and pressure sores.
Nitric oxide (NO). Although not proven to affect
survival, inhaled NO improves oxygenation and can be
used in life-threatening hypoxemia unresponsive to
other maneuvers. NO can be used safely in patients
who have increased ICP and in situations where it is
not possible to turn the patient to the prone
position.
Diuresis and fluid restriction. Regardless of
origin, diuresis and fluid restriction are
beneficial in ALI/ARDS but may be impossible to
achieve in patients who have vasospasm after SAH and
require hypertensive hypervolemic hemodilution
(triple-H) therapy.
Glucocorticosteroids. Moderate doses of glucocorticosteroids in the late phase of hypoxemic
ARDS may suppress fibroproliferation in the lung and
could be beneficial in patients who do not have
signs of infection.
Cardiogenic pulmonary edema. Pulmonary edema of
cardiogenic etiology can occur in the neurosurgical
patient. Left ventricular dysfunction may be from
myocardial ischemia or infarction, adverse drug
reactions, tachyarrhythmias, severe hypertension, or
fluid overload. Older patients are at greater risk;
however, myocardial ischemia from severe anemia,
hypotension, fluid overload, excessive mannitol
administration, and triple-H therapy can precipitate
pulmonary edema in the younger patient. Treatment
involves diuresis, fluid restriction, and supportive
care.
Pulmonary embolism. Pulmonary emboli are common in
critically ill neurosurgical patients. In SCI, the
incidence of deep vein thrombosis reaches 100%
without prophylaxis and 10% to 25% with prophylaxis.
Pulmonary emboli occur in >10% of SCI patients and
usually originate from venous thrombosis in lower
extremities and, less commonly, from either the
pelvic or prostatic venous plexi or the right
ventricle. Pulmonary embolism causes acute pulmonary
hypertension, right ventricular failure, and a
reduction in cardiac output. The diagnosis of
smaller pulmonary emboli should be considered in
patients who have unexplained tachypnea and chest
pain. The Pa O2 is usually normal, and the Paco2 is
decreased in the tachypneic patient who has small
pulmonary emboli. Massive pulmonary embolism causes
acute cardiorespiratory failure. In the past decade,
the multidetector row spiral CT scan has become the
first-line diagnostic tool for the diagnosis of
pulmonary embolism. If the multidetector row spiral
CT scan is not available, CT scan and pulmonary
angiography are indicated. Definitive treatment is
predominantly supportive and involves
anticoagulation. If anticoagulation is
contraindicated, an inferior vena cava filter should
be placed.
II. Respiratory care
Administration of oxygen and airway management. All
TBI patients should be considered hypoxic until
proven otherwise, and supplemental oxygen (O2)
should be administered. A Pa O2 of >60 mm Hg is
necessary to maintain adequate cerebral oxygenation
in patients after TBI and SCI.
Endotracheal intubation is indicated in the
neurosurgical patient for the following conditions:
Inability to protect the airway or clear secretions
Need to reduce ICP by control of ventilation
PaO2 <60 mm Hg in spite of supplemental O2 by mask
Paco2 >50 mm Hg
pH <7.2
Respiratory rate >40/minute or <10/minute
Tidal volume <3.5 mL/kg
Vital capacity <10 to 15 mL/kg
Muscle fatigue
Airway compromise
Hemodynamic instability
Orotracheal intubation. Orotracheal intubation is
preferable for urgent intubation in the presence of
increased ICP, hypoxemia, and/or hemodynamic
instability. In the hemodynamically stable patient,
either thiopental, 3 to 5 mg/kg, or propofol, 2.5
mg/kg, and succinylcholine, 1.5 mg/kg, can be used
to prevent coughing and an increase in ICP.
Alternatively, rocuronium, 1 to 1.5 mg/kg, can be
used for muscle paralysis. Etomidate, 0.1 to 0.3
mg/kg and lidocaine, 1 to 1.5 mg/kg, can be used in
the less stable patient. With patients who have a
full stomach, a modified rapid sequence induction
with cricoid pressure and ventilation by mask might
be used to avoid marked increases in ICP from
hypoxemia and hypercarbia. The head and neck should
be stabilized by an assistant (manual in-line
stabilization) to prevent head movement in the
presence of potential cervical spine injury. Manual
in-line stabilization may make a difficult
intubation more difficult and result in more
movement of unstable structures lower in the neck.
Nasotracheal intubation.
Nasotracheal intubation is
of minimal use in NICU patients because of the
possibility of resultant obstruction of sinus
drainage, which increases the risk of sinusitis,
pneumonia, and subsequent sepsis and ALI/ARDS.
Nasotracheal intubation is absolutely
contraindicated in the presence of a basilar skull
fracture. Awake nasotracheal intubation might
increase ICP in the head-injured patient.
Tracheotomy is absolutely indicated in the presence
of high SCI (higher than C5). Emergency tracheotomy
or cricothyrotomy could be required in patients who
have TBI and multiple facial fractures rendering
bag-and-mask ventilation difficult. Compared with
endotracheal intubation, tracheotomy decreases
anatomical dead space and the work of breathing
slightly, improves access for suctioning, provides
more comfort to the patient, and reduces sedation
requirements, all of which may be beneficial to
patients who have difficulties in weaning from the
ventilator. Early tracheotomy is considered in the
first week of admission. Although there is no
consensus as to the beneficial effects of early
surgical tracheotomy, performing a bedside
percutaneous dilatational tracheotomy (PDT) on the
second or third day after injury significantly
reduces the incidence of pneumonia, the ICU length
of stay, and the mortality in patients who were
expected to need mechanical ventilation for >14
days. Therefore, early bedside PDT should be
considered in patients who have significant
neurologic injury, absent gag reflex, or inability
to protect their airway and clear secretions and for
patients who are in a persistent vegetative state.
Because of the advantages of bedside PDT, including
negation of the need to involve the operating room
and to transport a critically ill patient, this
technique has gained popularity in recent decades.
The procedure requires the participation of two
physicians (surgeon, otolaryngologist, intensivist)
and consists of cricothyroidotomy and tracheal
dilatation using the Seldinger technique with
bronchoscopic guidance through the endotracheal tube
by an experienced bronchoscopist. Although
complications are rare with an experienced team, the
major complication is bleeding (1% to 2%).
Invasive mechanical ventilation is used to improve
gas exchange after nasal or oral endotracheal
intubation. Vital signs should be monitored and
arterial blood gases measured 30 minutes after
changing ventilator settings.
Controlled mechanical ventilation is used in the
operating room and occasionally in the NICU in
selected patients who have no ventilatory efforts
owing to neuromuscular blockade, high cervical SCI,
absent ventilatory drive, drug overdose, or brain
death.
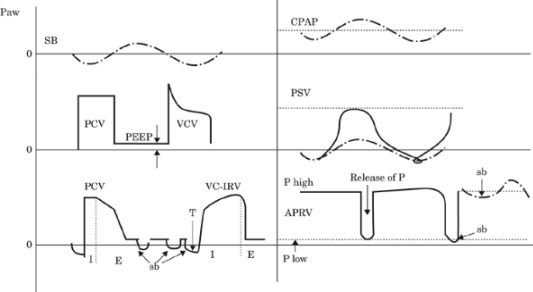
For patients in whom spontaneous ventilation is
preserved, the preferred assisted modes of
ventilation are those in which the ventilator
delivers a breath triggered by the patient's
inspiratory effort (Fig.-4). Along with classic
volume-controlled and pressure-controlled modes of
mechanical ventilation (assist-controlled,
synchronized and nonsynchronized intermittent
mandatory ventilation, and pressure support
ventilation), modern ventilators provide a variety
of new ventilation modes. These modes include "dual-controlled"�
mechanical ventilation with preset tidal volume and
inspiratory pressure ("volume-assured pressure
support"� or "pressure augmentation"� mode),
airway-pressure release ventilation (the mode of
pressure-controlled inverse-ratio ventilation,
allowing spontaneous breathing), and proportional
assist ventilation.
PEEP is used to increase arterial oxygenation by
alveolar recruitment and increasing FRC, which
decreases intrapulmonary shunting and [V with dot
above]A/[Q with dot above] abnormalities. The
application of PEEP decreases the need to give high,
potentially toxic, concentrations of oxygen. On the
other hand, PEEP increases Paco2 by increasing dead
space ventilation. Hemodynamically, PEEP decreases
venous return, cardiac output, and systemic arterial
blood pressure. These effects can be partially
attenuated by hydration. The varied clinical
responses of TBI patients to PEEP might be secondary
to PEEP's effect on other hemodynamic and
respiratory variables. The use of 10 cm H2O of PEEP
improves oxygenation but can decrease cerebral
perfusion from the decrease in cardiac output. PEEP
usually causes clinically inconsequential increases
in ICP in patients who have severe TBI. The
influence of PEEP on ICP is less prominent in
patients who have stiff lungs (as with ALI/ARDS) and
are presumably the ones who need PEEP the most. A
PEEP of up to 30 cm H2O has been used to treat
hypoxemia with minimal adverse effect on ICP.
However, because significant potentially serious
increases in ICP and clinical deterioration have
been observed in some TBI patients after the
application of PEEP, ICP should be monitored when
PEEP is used. Increases in ICP from PEEP can be
reduced by elevating the head 30� and concurrently
administering mannitol. The abrupt removal of PEEP
might also increase ICP.
|
Figure-4. Modes of
mechanical ventilation. |
 |
|
Paw,
airway pressure; SB, spontaneous breathing; PCV,
pressure-controlled ventilation; PEEP, positive
end-expiratory pressure; VCV, volume-controlled
ventilation; VC-IRV, volume controlled-inverse ratio
ventilation; I, inspirium; E, expirium; sb,
spontaneous breath; T, trigger; CPAP, continuous
positive airway pressure; PSV, pressure support
ventilation; APRV, airway pressure release
ventilation. |
Noninvasive mechanical ventilation is
contraindicated for neurosurgical patients in acute
respiratory failure who do not have an endotracheal
tube. This modality is useful only for patients who,
in the absence of neurologic deficits, have, for
example, an exacerbation of chronic obstructive
pulmonary disease.
Monitoring patients receiving mechanical
ventilation. The efficacy and safety of
mechanical ventilation in patients is evaluated by
monitoring gas exchange, airway pressure, breathing
pattern, lung compliance, hemodynamic function,
chest radiographs, and chest CT scans, the "gold
standard"� for diagnosis of lung pathology.
Weaning. Clinically stable patients should be
frequently reevaluated for readiness for a trial of
weaning. However, standard weaning criteria,
including normal neurologic status, are not
applicable to head-injured patients, in whom
impaired neurologic status can delay weaning and
extubation.
Clinical parameters to be met before attempting
weaning
1.
Cause for instituting mechanical ventilation is
sufficiently resolved.
2.
Adequate oxygenation: Pa O2/Fio2
≥ 150 to 200 or
hemoglobin saturation (SaO2) of >90% on Fio2 ≤ 0.4
to 0.5 with PEEP ≤ 8 cm H2O, pH ≥ 7.25.
3.
Hemodynamic stability (low-dose
inotropes/vasopressors are allowed).
Weaning techniques. Spontaneous breathing trial for
30 to 120 minutes should be attempted in patients
considered candidates for weaning from mechanical
ventilation. The trial can be performed with a
T-piece, using a low level of continuous positive
pressure (5 cm H2O) or pressure support (5 to 7 cm
H2O).
|
